ANOMALÍAS CONGÉNITAS
Se presentan en el 20% de todos los individuos.
ANOMALÍAS DE CANTIDAD DE TEJIDO
· Agenesia: es la falta de riñón, si es bilateral es incompatible con la vida. Unilateral: el único riñón compensa la función.
· Hipoplasia: poco desarrollo, si es bilateral lleva a la insuficiencia en la infancia; y si es unilateral el otro compensa.
· Supernumerarios.
ANOMALÍAS DE POSICIÓN
· Ectopía: estasis, puede estar dentro de la pelvis con uréteres tortuosos, dificulta la salida de orina, predispone a infecciones.
· Riñón en herradura: se fusionan ambos riñones por uno de sus polor (en el 90% de los casos es el inferior).
ANOMALÍAS DE DIFERENCIACIÓN (QUISTES)
· Displasia quística renal: aumento del tamaño renal, si es bilateral lleva a la insuficiencia. Hay anomalías de la diferenciación, múltiples quistes con islotes de mesénquima intermedio (patognomónico). Hay nefronas normales junto a nefronas inmaduras. Los quistes se recubren de epitelio plano y contienen líquido seroso.
· Enfermedad poliquística renal:
1. Del adulto: siempre es bilateral, riñones poliquísticos, que borran los detalles normales por compresión y atrofia, pesan hasta 4 kilos cada uno. El epitelio de los quistes es plano y contienen un líquido seroso-hemorrágico. Los quistes se ven desde la superficie externa. La falla está en los genes de la policisteína 1 (85% de los casos) o 2 (15% restante), (PDK1 y PDK2) complejos de proteínas de membrana del epitelio de los túbulos que regula la secreción de los mismos, en este caso la secreción de líquido es excesiva, lo que provoca la dilatación de los túbulos formando quistes primero miscroscópicos que se presentan en la infancia, pero en edad adulta son macroscópicos y llevan a la insuficiencia renal alrededor de los 50 años. Estas proteínas participan en la interacción célula-célula y célula-matriz, interacciones necesarias para regular la proliferación y maduración del epitelio. Puede haber quistes en otros sectores: hígado, bazo, páncreas. La mutación de PKD2 tarda más en llegar a la IRC.
2. Del niño: semejante a la anterior, pero lleva a la insuficiencia de la infancia y los quistes macroscópicos no se ven desde la superficie externa, la cual es lisa. La falla está en el gen de la fibrocistina (PKHD1), proteína de membrana que actúa como receptor necesario para la diferenciación de los túbulos colectores y de los conductos biliares.
· Enfermedades quísticas de la médula renal:
1. Riñón en esponja medular: dilatación quística de los colectores medulares. La etiología es desconocida, se manifiesta en adultos y no trae consecuencias.
2. Nefroptisis/enfermedad quística medular urémica: enfermedad renal que comienza en la infancia y puede llevar a la insuficiencia renal en edad adulta. Hay quistes medulares con atrofia cortical tubular y daño túbulointersticial, se conservan los glomérulos. Riñones pequeños con quistes medulares. La nefronoptisis parece ser la causa genética más frecuente de IR en niños y jóvenes. Falla de nefrocistinas, proteínas de epitelio de túbulos de función desconocida.
· Quistes adquiridos asociados a diálisis: quistes en corteza y médula, con epitelio plano, contenido seroso y cristales de oxalato de calcio. Se formarían por obstrucción de los túbulos (fibrosis o calcio), pueden sangrar. Peligro: se puede originar un carcinoma a partir del epitelio de los quistes, el riesgo es del 7% después de 10 años.
· Quistes simples: únicos o múltiples, de 1 a 5 cm., contiene líquido claro, epitelio plano en única capa, y se rodean externamente por fibrosis. Se ubican en corteza y no dan síntomas, la importancia es no confundirlos con tumores (los quistes son líquidos, los tumores sólidos).
GLOMERULONEFRITIS O GLOMERULOPATÍAS
Procesos inflamatorios que afectan a los glomérulos y pueden llevar o no a la disfunción de los mismos, cuando no hay componente inflamatorio se llaman glomerulopatías.
· Son de dos tipos: primarios (se afectan los riñones en forma exlusiva) y secundarios (los riñones se afectan en el curso de una enfermedad sistémica).
· Alteraciones histológicas: proliferación celular, infiltrado leucocitario, engrosamiento de la membrana basal glomerular, esclerosis o hialinización.
· Patogenia: mecanismos inmunológicos:
1. Por anticuerpos: por formación de complejos inmunes in situ (tipo II), y por depósito de complejos inmunes (tipo III).
2. Por células: linfocitos T.
3. Por vía alterna del complemento (C3).
Mecanismos de progresión del daño glomerular:
1. Pérdida del polianión glomerular: proteinuria.
2. Hiperfiltración y cambios hemodinámicos.
3. Fibrosis tubulointersticial.
· Conceptos útiles:
Difuso: afecta a todos los glomérulos.
Focal: afecta a un grupo de glomérulos.
Global: afecta todo un glomérulo.
Segmentario: afecta una parte del glomérulo.
Mesangial: afecta preferentemente al mesangio.
GLOMERULONEFRITIS PRIMARIAS
Clasificación según síndromes:
· Síndrome nefrítico: GNF proliferativa aguda (o postestreptocócica). Hipertensión, hematuria macroscópica, azotemia. Proteinuria y edema leve a moderado.
· Síndrome de GNF rápidamente progresiva (semilunas): tipo I (idiopática y S. de Goodpasture), tipo II (idiopática, postinfecciosa), tipo III (idiopática, granulomatosis de Wegener). Hematuria, proteinuria y oliguria grave que lleva a la IRA en semanas.
· Síndrome nefrótico: GNF membranosa (se forman espigas), nefrosis lipoidea (desaparecen los pedicelos), glomeruloesclerosis focal y segmentaria (alteraciones inespecificas), GNF membranoproliferativa (vías de tren en la MBG). Proteinuria mayor a 3,5 grs. por día (rango nefrótico), hipoalbuminemia, edemas, hiperlipidemia como reacción a la pérdida de proteínas, lipiduria, predisposición a infecciones por la pérdida de anticuerpos y factores del complemento, y es un estado de hipercoagulabilidad por la pérdida de anticoagulantes por orina.
· Síndrome de insuficiencia renal crónica: es la claudicación de un riñón insuficiente luego de un largo período de enfermedad. Comienza con azotemia (elevación de la creatinina y del nitrógeno ureico en sangre por un descenso del filtrado glomerular) y cuando el filtrado es menor al 20% aparece la uremia (hiperazoemia más un conjunto de síntomas, signos y alteraciones bioquímicas). GNF crónica (estadío final de todas las GNF que evolucionan, aunque la cuarta parte llega directamente).
Alteraciones clínicas:
1. En el volumen y en el pH: al descender el filtrado se acumula NA y hay aumento de la volemia, pero también se retienen metabolitos ácidos, lo que lleva a una acidosis metabólica.
2. En el metabolismo de Ca y hueso: riñón enfermo no sintetiza vitamina D3, y no se puede absorber Ca, entonces reacciona la paratiroides, hiperparatiroidismo secundario, con un aumento de PTH, que remueve Ca de los huesos, esto predispone a retrasos en el crecimiento y a fracturas patológicas.
3. Cardiopulmonares: el aumento de la volemia puede llevar a una insuficiencia cardíaca y a riesgo de edema de pulmón, también puede haber pleuritis fibrinosa.
4. Hematopoyéticas: hay anemia por disminución de la síntesis de eritropoyetina, y puede haber diatesis (predisposición) hemorrágicas porque se altera la adhesión plaquetaria.
5. Gastrointestinales: esofagitis y gastroenterocolitis.
6. Dermatológicas: color cetrino de la piel (amarillo limón), por el depósito de urocromo (pigmento urinario) que se acompaña de prurito.
7. Neuromusculares. miopatías, convulsiones y coma.
La urea no es tóxica, si lo son los metabolitos acumulados que la acompañan.
· Hematuria o proteinuria asintomática: nefropatía por IgA o enfermedad de Berger.
GLOMERULONEFRITIS SECUNDARIAS
· Lupus (produce 5 lesiones): Patogenia: reacción de hipersensibilidad de tipo III (depósito de complejos inmunes) en glomérulos y capilares.
Produce 6 lesiones y ninguna es específica de lupus.
Tipo I: sin cambios o mesangial mínimo.
Tipo II: GNF. mesangial: hay escasa proliferación de células mesangiales y de matriz, escaso depósito de complejos inmunes en mesangio. Los capilares están respetados. Cursa con proteinuria o hematuria asintomática.
Tipo III: GNF proliferativa focal: proliferación focal y segmentaria de células mesangiales y del endotelio, hay necrosis fibrinoide de vasos. Infiltrado inflamatorio. Cursa con proteinuria o hematuria asintomática.
Tipo IV: GNF proliferativa difusa: es la más frecuente y la más grave. Proliferación difusa y global de células mesangiales, endotelio y epitelio (a veces este último prolifera y forma semilunas), con necrosis fibrinoide en capilares. Infiltrado inflamatorio. Cursa con síndrome nefrótico.
Tipo V: GNF membranosa: semejante a la primaria, cursa con síndrome nefrótico.
Los depósitos de complejos inmunes a nivel subendotelial producen lesiones en asa de alambre, que son típicos de las lesiones III y IV, mientras que en la de tipo V suelen ser subepiteliales.
Tipo VI: GNF esclerosante: es la lesión terminal, con esclerosis difusa y global.
· Púrpura de Henoch-Schonlein: se caracteriza por púrpuras en piel de brazos, piernas y nalgas; síntomas abdominales como dolor, hemorragia intestinal; artralgia no migratoria; alteraciones renales (GNF semejante a la nefropatía por IgA, se cree que serían la misma enfermedad. Es más frecuente en niños).
Patogenia: se forman complejos inmunes por IgA que se depositan en glomérulos y en los pequeños vasos de los tejidos afectados.
· Nefropatía diabética: conjunto de lesiones que aparecen en los riñones de un paciente diabético. La insuficiencia renal es la segunda causa de muerte y es más frecuente en la DBT tipo I. Es una de las principales causas de IRC. Las lesiones son:
1. Arteriolas: arterioloesclerosis hialina.
2. Túbulos: engrosamiento de la membrana basal.
3. Mayor predisposición a pielonefritis con el cuadro de necrosis papilar.
4. Glomérulos: 3 tipos de lesiones: engrosamiento de la membrana basal de los capilares (forma parte de la microangiopatía diabética, se inicia dos años después del comienzo de la DBT), glomeruloesclerosis mesangial difusa (se engrosa la MBG por depósitos de proteínas, y hay aumento de la matriz mesangial en forma global y difusa, con leve proliferación de células mesangiales), glomeruloesclerosis nodular o enfermedad de Kimmelstiel-Wilson (masas hialinas ovoides o esféricas que están en la periferia del glomérulo y se rodean de capilares. Su importancia radica en que es altamente específica de DBT. Se ven unos 10 a 20 años del inicio de la DBT).
Evolución: con el tiempo las masas crecen comprimiendo los capilares, lo que provoca isquemia, esto lleva a la fibrosis del glomérulo. También puede acompañarse de una lesión mínima llamada gota capsular y consiste en un acúmulo hialino en la cápsula de Bowman.
Patogenia: alteración de la MBG con pérdida del polianión glomerular a causa de la glucosilación no enzimática, cambios hemodinámicos e hiperfiltración.
Clínica: comienzan con microalbuminuria y con aumento del filtrado glomerular, luego aparece la proteinuria, primero leve, luego llega a rango nefrótico, aquí disminuye el filtrado glomerular y llegan a la insuficiencia renal en 5 años. El proceso puede llevar entre 20 y 25 años, el control adecuado de las glucemias previene o retrasa la afectación renal.
· Amiloidosis: depósito de amiloide a nivel mesangial y en la MBG a nivel subendotelial, con el tiempo oblitera los glomérulos. Se ve en la amiloidosis sistémica reactiva. Tinción: rojo congo. Cursan con síndrome nefrótico e insuficiencia renal.
· Vasculitis: poliarteritis microscópica y granulomatosis de Wegener.
· Endocarditis bacteriana: es por depósito de complejos inmunes, generalmente produce un síndrome nefrítico, más rara vez una GNF rápidamente progresiva.
GLOMERULONEFRITIS HEREDITARIAS
· Síndrome de Alport: generalmente ligada a X, presenta sordera, transtornos visuales (cataratas, alteración de la córnea) y afectación glomerular: glomeruloesclerosis difusa por la proliferación de la matriz.
Patogenia: existe un defecto hereditario en la síntesis del colágeno de tipo 4, no se sintetiza la cadena alfa5. Menos frecuentemente, en los casos autosómicos faltan las cadenas 3 o 4.
Clínica: los síntomas comienzan entre los 5 y 20 años de edad con hematuria micro o macroscópica, y llegan a la IRC entre los 20 y 50 años de edad.
· Enfermedad de la membrana fina (o hematuria benigna familiar): la MBG es más delgada que en las personas normales (la mitad), al ser más frágil puede producir hematuria asintomática. La función renal se conserva normal. El defecto está en los genes que codifican las cadenas alfa 3 o alfa 4 del colágeno de tipo 4.
IRA: supresión brusca de la función renal, es un proceso bilateral, puede ser reversible.
Etiologías:
1. Prerrenal (se da por hipovolemia): IC, shock, aterosclerosis.
2. Intrarrenal:
Por afectación de vasos: vasculitis, hipertensión, eclampsia.
Por afectación de glomérulos: GNF.
Por afectación de túbulos: necrosis tubular aguda (causa más frecuente de IRA).
3. Postrenal: es rara, se da por obstrucción ureteral bilateral por cálculos o trombos.
Clínica: azotemia, oliguria, síndrome urémico.
ENFERMEDADES TUBULARES
NECROSIS TUBULAR AGUDA
Principal causa de IRA. Isquémica (shock) y nefrotóxica (agente tóxico).
ENFERMEDADES TUBULOINTERSTICIALES
PIELONEFRITIS
· Aguda:
· Crónica:
1. Por reflujo:
2. Por obstrucción:
FÁRMACOS Y TOXINAS
· Nefritis intersticial aguda:
· Nefritis intersticial crónica:
POR TRANSTORNOS METABÓLICOS
· Nefropatía por uratos
1. Aguda:
2. Crónica:
3. Nefrolitiasis:
· Nefropatía por hipercalcemia
1. Cálculos.
2. Nefrocalcinosis:
NEOPLASIAS
Mieloma múltiple:
IRRADIACIÓN
HIPERTENSIÓN
Presión elevada y mantenida por encima de 90 mmHg. la diastólica y por encima de 140mmHg. la sistólica.
HIPERTENSIÓN PRIMARIA O ESENCIAL (90%)
Es de origen desconocido.
· Benigna:
· Maligna:
HIPERTENSIÓN SECUNDARIA (10%)
· Renales:
· Endócrinas:
· Cardiovascular:
· Neurógena:
Órganos blanco: cerebro (se pueden producir hemorragias), corazón (hipertrofia del VI e ICI), retina (ceguera), riñón (nefroesclerosis benigna o maligna), vasos (de mediano y gran calibre predispone a la aterosclerosis, y de pequeño calibre a la arteriolosclerosis).
INFARTO RENAL
Es relativamente frecuente porque recibe el 25% del gasto cardíaco. Causas:
1. Embolos: es la más frecuente.
2. Trombos asociados a placa de ateroma.
3. Vasculitis.
Son infartos blancos.
Clínica: cursan con dolor espontáneo costovertebral y hematuria.
MICROANGIOPATÍAS TROMBÓTICAS
SÍNDROME HEMOLÍTICO URÉMICO
· Típico:
· Atípico:
PÚRPURA TROMBÓTICA TROMBOCITOPÉNICA
NEFROLITIASIS
Cálculos en riñón. Están compuestos por calcio (75%), fosfato amónico-magnésico (15%), ácido úrico (6%), cistina (1 al 3%).
TUMORES
BENIGNOS
· Adenoma papilar renal: se origina en el epitelio de los túbulos renales y forma papilas. Están siempre en la corteza, son de color amarillos, la cápsula es incompleta y miden no más de 0,5 cm. Tienen la misma histología que el hipernefroma y no se los puede diferenciar desde el punto de vista histológico ni por pruebas inmunohistoquímicas. Hay una regla poco fiable que dice: si mide menos de 3 cm. es un adenoma, si mide más un hipernefroma.
· Angiomiolipoma: formado por vasos, músculo liso y grasa. Se caracteriza porque puede dar hematuria.
· Oncocitoma: formado por células eosinófilas, con núcleos pequeños y cargadas de mitcondrias. Se origina en las células intercaladas de los túbulos colectores. Está encapsulado y es de color bronce. En raros casos puede metastatizar.
· Hemangioma.
· Tumor de células yuxtaglomerulares: es raro, produce hipertensión secundaria por liberación de renina.
MALIGNOS
· Carcinoma de células renales o hipernefroma (o tumor de Grawitz): 90%. Existen de varios tipos:
1. Carcinoma de células claras no papilar (80%), es unilateral.
2. Carcinoma papilar (14%): son multifocales y bilaterales, y son los que más se asocian a diálisis. Pueden tener cuerpos de Psamoma. Células cúbicas eosinófilas, granulares.
3. Carcinoma renal cromófobo (5%): eosinófilas pálidas. Deriva de las células intercalares de los colectores. Es el de mejor pronóstico.
4. Carcinoma de los conductos colectores (1%): células epiteliales atípicas con aspecto de tachuelas.
· Nefroblastoma o tumor de Wilms: síndromes genéticos asociados con el cromosoma 11:
1. Síndrome WAGR (Wilms, Aniridia, Anomalías Genitales y Retraso mental), falla el gen WT-1.
2. Síndrome Denys-Drash, falla el gen WT-1. Disgenesia gonadal más nefropatía glomerular.
3. Síndrome de Beckwith-Wiedemann, falla el gen WT-2. Organomegalia (hipertrofia de órganos corporales).
lunes, 19 de agosto de 2013
domingo, 18 de agosto de 2013
PATOLOGÍAS RESPIRATORIAS II: RESTRICTIVAS Y TUMORES
PATOLOGÍAS RESTRICTIVAS DE PULMÓN
Son neumopatías que afectan al intersticio pulmonar, el cual se engrosa. Primero forma reticulados, luego nódulos y termina en pulmón en panal. Estos procesos comienzan con una alveolitis, es el acúmulo de células inflamatorias en intersticio y en los alveolos. La liberación de mediadores químicos produce lesión en el intersticio y fibrosis. Se dividen en fibrosantes, granulomatosas, esosinofilicas, por tabaco, hemorrágica y otras.
FIBROSANTES
Neumoconiosis
Presencia de polvo orgánico o inorgánico en el pulmón, que origina una reacción pulmonar al mismo (colagenización). La colagenización comienza siendo nodular y si progresa se hace difusa.
Para que se produzca hay que tener en cuenta el tipo de polvo, tamaño de las partículas (las medianas son las más peligrosas), cantidad retenida y asociación con otros irritantes como el tabaco.
Las más importantes son:
1. Neumoconiosis de los trabajadores de carbón: es benigna y presenta 2 formas:
· Leve o simple: es la más frecuente, se da en mineros de carbón con más de 10 años de exposición. Comienza como un cuadro de antracosis pulmonar: oscurecimiento de los pulmones que se produce en fumadores y habitantes de ciudades, consiste en la presencia de macrófagos cargados de polvo de carbón alrededor de bronquiolos respiratorios. Esto no trae consecuencias, pero en los mineros progresa hacia la formación de máculas de carbón de 1 cm. de diámetro, que se fibrosan formando nódulos de hasta 2 cm. (colagenización nodular). Clínica: tos y esputo negruzco.
· Grave o compleja: (colagenización difusa) se da por la progresión de algunos casos de la forma leve. Los nódulos se hacen confluentes, y forman áreas de hasta 10 cm. que en el centro tienen un líquido semejante a la tinta china. Lleva a la insuficiencia respiratoria o al core pulmonale. No hay relación con cáncer de pulmón pero si puede asociarse a enfisema y a bronquitis crónica.
2. Silicosis: acumulación de polvo de sílice (como el cuarzo), se encuentra en minas de oro, estaño, cobre, en la industria de la cerámica, mármol, vidrio, chorro de arena. Tiene 2 etapas: nodular (nódulos pequeños y aislados) y difusa (desarrolla patrón en panal, por colagenización difusa). Se puede complicar con tuberculosis (la predispone porque altera la función de los linfocitos Th1).
3. Asbestosis: es la acumulación de asbesto o amianto, se encuentra en tuberías de agua, alcantarillados, tejados, cubierta de frenos, envoltura de embragues, como material aislante. Las partículas de asbesto son muy largas y livianas, los macrófagos las fagocitan y forman unas estructuras con los extremos en forma de masa y están envueltos con hemosiderina, se llaman cuerpos ferruginosos. El proceso comienza en los lóbulos inferiores, las anteriores en los superiores. Son características las placas pleurales, es colágeno denso con calcificaciones en la pleura parietal. Lleva a la fibrosis intersticial difusa o a cáncer. Aumenta la frecuencia de mesotelioma maligno 1000 veces, de cáncer de pulmón unas 5 veces (y si además el individuo fuma la aumenta 55 veces), carcinoma de laringe e hipernefrona.
4. Beriliosis: acumulación de berilio, lo encontramos en la industria espacial, energía nuclear, electrónica. Puede ser:
· Aguda: es por exposición corta, pero intensa (dosis elevadas). Puede resolverse u organizarse. Cursa con la patogenia general.
· Crónica: se da por exposición lenta y prolongada. Se forman granulomas similares a los de la sarcoidosis, con los cuerpos de Schauman y los cuerpos asteroides. Patogenia: reacción de hipersensibilidad tipo 4, existe cierta predisposición genética.
Aumenta 2 veces el riesgo de cáncer de pulmón.
Otras neumoconiosis: bisinosis (inhalación de fibras de algodón). Cualquiera de estas neumoconiosis se puede complicar con nódulos reumatoideos en pulmón, ésta asociación se llama Síndrome de Caplan. Posibles causas de muerte: cáncer, insuficiencia respiratoria, cor pulmonale.
Fibrosis pulmonar idiopática (Síndrome de Haman Rich)
Es más frecuente en varones adultos. Es un proceso destructivo de los tabiques alveolares de origen desconocido que lleva a la fibrosis intersticial difusa con el cuadro de pulmón en panal. El agente responsable no se ha podido identificar, pero produce episodios repetidos de lesión aguda que culminan en la fibrosis. Los linfocitos Th2 colaboran con la inflamación. Los neumocitos tipo I liberan TGF-beta, y este parece ser el motor del proceso, como estímulo fundamental de fibroblastos. La fibrosis es subpleural y sigue los septos interlobulillares. No hay cambios morfológicos más allá de la fibrosis. Por esto último, por se un proceso idiopático y porque puede verse en otros procesos, el diagnóstico se hace por descarte. Clínica: hipoxemia, cianosis grave, lleva a la muerte por insuficiencia respiratoria en 3 años. Tratamiento: trasplante de pulmón.
Neumonía organizativa criptogenética
Etiología desconocida, se producen formaciones fibrosas polipoideas dentro de alvéolos y bronquiolos (cuerpos de Masson), que en la radiografía se ven como áreas de condensación. No produce pulmón en panal.
Clínica: disnea, tos, se recuperan con corticoides en tratamiento durante 6 meses.
Fármacos, irradiación
Autoinmunes: lupus, artritis reumatoidea, esclerodermia
GRANULOMATOSAS
Neumonitis por hipersensibilidad o alveolitis alérgica
Se da por inhalación de polvos orgánicos, como antígenos de plumas, de excrementos de aves, de esporas bacterianas, de esporas de hongos.
· Patogenia: 6 hs. después de la esposición se produce una reacción de hipersensibilidad de tipo III, con depósito de complejos inmunes en las paredes de los pequeños vasos pulmonares del intersticio. También hay reacción de tipo IV, con formación de granulomas en intersticio.
· Morfología: hay inflamación intersticial, fibrosis, granulomas.
· Clínica: 6 hs. después de la exposición presentan fiebre, tos, disnea y leucocitosis.
· Ejemplos:
1. El pulmón del granjero: es por exposición al heno almacenado húmedo, donde proliferan actinomicetos.
2. El pulmón del cuidador de palomas: por inhalación de antígenos de plumas o excrementos de aves.
3. El pulmón del aire acondicionado: por bacterias termófilas presentes en agua caliente.
Sarcoidosis
EOSINOFÍLICAS
Eosinofilia pulmonar
El ejemplo es el Síndrome de Loeffler. Son áreas de condensación cambiantes de eosinófilos a nivel pulmonar, desencadenadas por la presencia de parásitos como Ascaris lumbricoides. Hay liberación aumentada de IL5.
POR TABACO
Neumonitis intersticial descamativa (NID)
Es más frecuente en varones entre 40 y 50 años, es rara.
· Morfología: hay aumento de macrófagos en los alvéolos, que contienen un pigmento pardo grisáceo (macrófago del fumador) con escasos neumocitos descamados, rara vez se produce una fibrosis intersticial difusa que lleva a la insuficiencia respiratoria. Buen pronóstico si abandonan el tabaco.
Enfermedad pulmonar intersticial asociada con bronquiolitis respiratoria
Macrófagos semejantes a la NID, a nivel de bronquiolos respiratorios, puede progresar hacia la fibrosis.
HEMORRÁGICAS
Síndrome de Goodpasture
Enfermedad autoinmune donde se producen anticuerpos contra la membrana basal glomerular y contra los tabiques alveolares (especificamente contra el colágeno de tipo 4, cadena alfa3). Hay predisposición genética en relación con los HLA. Es más frecuente en varones de 20 a 30 años.
· Clínica: produce hemoptisis y una glomerulonefritis rápidamente progresiva (esta última es la principal causa de muerte, lleva a insuficiencia renal).
· Tratamiento: inmunosupresores y plasmaféresis (recambio de plasma).
Vasculitis
OTRAS
Proteinosis alveolar pulmonar
Dentro de los alvéolos se acumula un material semejante al surfactante, es rico en proteínas y fosfolípidos. Se acompaña de macrófagos y algunos neumocitos necróticos.
· Patogenia: defecto en el aclaramiento de los macrófagos.
· Hay 3 variantes:
1. Adquirida (90%): se da en adultos, hay carencia del factor de crecimiento hematopoyético (GM-CSF) por autoanticuerpos contra el mismo. Generalmente no progresa hacia la fibrosis intersticial. Clínica: tos, disnea, esputo (material gelatinoso) que da riesgo de infecciones.
2. Congénita: donde hay déficit del factor o del receptor para el GM-CSF, o mutaciones en proteínas del surfactante. Lleva a la muerte 3 a 6 meses luego del nacimiento.
3. Secundaria: rara, se asocia a neoplasias, inmunodeficiencias.
PLEURA
PLEURITIS
· Serosa: Por infecciones virales, no dejan secuelas.
· Fibrinosa: por lupus, uremia, artritis reumatoidea, radioterapia. Se resuelve o se organiza.
· Supurada (empiema): es por la llegada de bacterias u hongos al espacio pleural. Las vías de llegada son desde un foco pulmonar, por vía hemática, o desde un absceso subdiafragmático. Generalmente se organizan, dejando secuelas graves (adherencias).
· Hemorrágica: se da por neoplasias pleurales, infecciones por rickettsias. Secuelas graves.
TUMORES
· Mesotelioma benigno o tumor fibroso solitario: es un fibroma, se lo considera un tumor de tejidos blandos, se conecta a la pleura por un pedículo. Células semejantes a fibroblastos, CD 34 +, queratina -.
· Mesotelioma maligno: se asocia al asbesto y produce derrame pleural. Se origina en pleura visceral o parietal. El VS40 (virus de los simios 40) podría tener relación (inactivaría al P53 y RB). Tienen 3 variantes:
1. Sarcomatoide o mesenquimal (20%): se parece a un fibrosarcoma, son células fusiformes.
2. Epitelial (60%): CD 34 -, queratina +. Forma papilas, es dificil distinguirlo de un adenocarcinoma bronquioloalveolar.
3. Mixto (20%): combinación de los 2 anteriores. Producen una afectación difusa. Invaden pulmón, ganglios regionales y metástasis al hígado. Clínica: dolor torácico, disnea, derrame pleural recurrente. Mueren al año del diagnóstico.
LARINGE
LARINGITIS
Se producen por:
· Crup: el bacilo diftérico produce una toxina que genera la formación de una seudomembrana en la mucosa laringea, ésta tiende a desprenderse, siendo grave en niños pequeños por el escaso calibre de la laringe, pudiendo llevar a la muerte por asfixia.
· Seudocrup: lo producen los virus, las secreciones serosas se depositan en las cuerdas vocales, se deshidratan y general una disnea inspiratoria con el estridor laringeo. La obstrucción generalmente es parcial y no llega a la asfixia.
TUMORES
· Polipos laringeos: nódulos redondos, sésiles o pediculados, miden hasta 1 cm., se ubican en las cuerdas vocales. Se asocian a tabaco, someter la voz a grandes esfuerzos (maestras, cantantes, se lo llama el nódulo de los cantantes). Están formados por un eje de tejido conectivo rodeado de epitelio escamoso. Clínica: ronquera progresiva. En los cantantes es bilateral, en el resto unilateral.
· Papiloma laringeo: se parece a una frambuesa, mide hasta 1 cm. Se ubica en las cuerdas vocales. Son varios ejes digitiformes de tejido conectivo rodeados de epitelio escamoso. Se producen por HPV 6 u 11. Si aparece en niños pequeños suele ser una papilomatosis, y el contagio se produjo por aspiración en el canal del parto desde lesiones de condoloma acuminado materno. Clínica: ronquera, si son múltiples puede obstruir la vía aérea.
· Carcinoma epidermoide (maligno, 95%): es más frecuente en varones mayores de 40 años. Predisponentes: tabaco, asbesto, infección por HPV, alcohol, irradiación. Surgen a partir de hiperplasias atípicas y displasia previa.
Morfología: pueden ser intrínsecos (crecen hacia el interior de la laringe) o extrínsecos (crecen hacia afuera, invadiendo estructuras vecinas). Según la ubicación pueden estar:
1. Por arriba de las cuerdas vocales, esto genera disfagia.
2. En las cuerdas vocales, son los más frecuentes y los que tienen bajo riesgo de metástasis porque están bien diferenciados y por la escasa presencia de linfáticos en las cuerdas. Generan disfonía.
3. Por debajo de las cuerdas vocales, generan disnea.
Evolución: metastatizan a ganglios regionales del cuello. La sobrevida a los 5 años es del 50%.
CAVIDADES NASALES Y SENOS PARANASALES. FARINGE
SINUSITIS
Es la inflamación de los senos paranasales. Surge de una rinitis complicada. La rinitis produce obstrucción de un meato, esto es una circunstancia de estasis, que predispone al ingreso de bacterias hacia el seno, cargándose de pus. Puede haber complicaciones, en el caso del seno maxilar puede alcanzar la órbita, el resto de los senos pueden producir osteomielitis y desde allí meningitis. El seno maxilar se puede comprometer a partir de una infección periapical desde la boca, en este caso aparece una flora infecciosa mixta proveniente de la misma. Existe una asociación poco frecuente con el Síndrome de Kartagener.
FARINGITIS
En el 90% son de origen viral (los mismos virus de las rinitis), en el 10% son de origen bacterianas por estafilococo y estreptococo beta hemolítico del grupo A (existe relación con fiebre reumatoidea y glomerulonefritis postestreptocócica).
TUMORES
· Angiofibroma nasofaringeo: se da en jóvenes, más frecuente en varones, formado por abundantes vasos, su característica es ser muy sangrante, pudiendo llevar a la muerte por la hemorragia.
· Papiloma invertido: crece al revés, hacia el hueso, infiltrando. Para curarlo hay que extirparlo completamente. Suele aislarse a los HPV 6 u 11. Histología: epitelio escamoso o cilíndrico. Otras variantes de papilomas son el septal (que es el más frecuente) y cilíndrico, ambas variantes son exofíticas.
· Carcinoma nasofaringeo (maligno): de células escamosas queratinizante, de células escamosas no queratinizante (EVB) e indiferenciado (EVB).
Se ubica más frecuentemente en el cavum (el techo de la faringe). Puede haber asociación con el EBV. Otros predisponentes son el tabaco y las nitrosaminas. En Africa son más frecuentes en niños, pero en China son más frecuentes en adultos. Diseminan a ganglios cervicales. La sobrevida a los 3 años es del 60%.
· Linfoma de células NK/T, de tipo nasal (granuloma letal de la línea media, maligno): tiene infecciones bacterianas añadidas con formación de granulomas y ulceraciones. Se creía que era una inflamación crónica de mal pronóstico por su penetración, hoy se sabe que es un linfoma.
· Neuroblastoma olfatorio (maligno): estesioneuroblastoma, se origina en las células neuroendócrinas dispersas (sinaptofisina, CD56, enolasa neuronal específica y cromogranina +) en la mucosa olfatoria. Son células redondas pequeñas semejantes a neuroblastos. Poseen gránulos secretores. Son ampliamente metastatizantes. Sobrevida a los 5 años del 60%.
· Plasmocitoma (o mieloma solitario): forma polipoide, generalmente no se transforma en mieloma múltiple.
Son neumopatías que afectan al intersticio pulmonar, el cual se engrosa. Primero forma reticulados, luego nódulos y termina en pulmón en panal. Estos procesos comienzan con una alveolitis, es el acúmulo de células inflamatorias en intersticio y en los alveolos. La liberación de mediadores químicos produce lesión en el intersticio y fibrosis. Se dividen en fibrosantes, granulomatosas, esosinofilicas, por tabaco, hemorrágica y otras.
FIBROSANTES
Neumoconiosis
Presencia de polvo orgánico o inorgánico en el pulmón, que origina una reacción pulmonar al mismo (colagenización). La colagenización comienza siendo nodular y si progresa se hace difusa.
Para que se produzca hay que tener en cuenta el tipo de polvo, tamaño de las partículas (las medianas son las más peligrosas), cantidad retenida y asociación con otros irritantes como el tabaco.
Las más importantes son:
1. Neumoconiosis de los trabajadores de carbón: es benigna y presenta 2 formas:
· Leve o simple: es la más frecuente, se da en mineros de carbón con más de 10 años de exposición. Comienza como un cuadro de antracosis pulmonar: oscurecimiento de los pulmones que se produce en fumadores y habitantes de ciudades, consiste en la presencia de macrófagos cargados de polvo de carbón alrededor de bronquiolos respiratorios. Esto no trae consecuencias, pero en los mineros progresa hacia la formación de máculas de carbón de 1 cm. de diámetro, que se fibrosan formando nódulos de hasta 2 cm. (colagenización nodular). Clínica: tos y esputo negruzco.
· Grave o compleja: (colagenización difusa) se da por la progresión de algunos casos de la forma leve. Los nódulos se hacen confluentes, y forman áreas de hasta 10 cm. que en el centro tienen un líquido semejante a la tinta china. Lleva a la insuficiencia respiratoria o al core pulmonale. No hay relación con cáncer de pulmón pero si puede asociarse a enfisema y a bronquitis crónica.
2. Silicosis: acumulación de polvo de sílice (como el cuarzo), se encuentra en minas de oro, estaño, cobre, en la industria de la cerámica, mármol, vidrio, chorro de arena. Tiene 2 etapas: nodular (nódulos pequeños y aislados) y difusa (desarrolla patrón en panal, por colagenización difusa). Se puede complicar con tuberculosis (la predispone porque altera la función de los linfocitos Th1).
3. Asbestosis: es la acumulación de asbesto o amianto, se encuentra en tuberías de agua, alcantarillados, tejados, cubierta de frenos, envoltura de embragues, como material aislante. Las partículas de asbesto son muy largas y livianas, los macrófagos las fagocitan y forman unas estructuras con los extremos en forma de masa y están envueltos con hemosiderina, se llaman cuerpos ferruginosos. El proceso comienza en los lóbulos inferiores, las anteriores en los superiores. Son características las placas pleurales, es colágeno denso con calcificaciones en la pleura parietal. Lleva a la fibrosis intersticial difusa o a cáncer. Aumenta la frecuencia de mesotelioma maligno 1000 veces, de cáncer de pulmón unas 5 veces (y si además el individuo fuma la aumenta 55 veces), carcinoma de laringe e hipernefrona.
4. Beriliosis: acumulación de berilio, lo encontramos en la industria espacial, energía nuclear, electrónica. Puede ser:
· Aguda: es por exposición corta, pero intensa (dosis elevadas). Puede resolverse u organizarse. Cursa con la patogenia general.
· Crónica: se da por exposición lenta y prolongada. Se forman granulomas similares a los de la sarcoidosis, con los cuerpos de Schauman y los cuerpos asteroides. Patogenia: reacción de hipersensibilidad tipo 4, existe cierta predisposición genética.
Aumenta 2 veces el riesgo de cáncer de pulmón.
Otras neumoconiosis: bisinosis (inhalación de fibras de algodón). Cualquiera de estas neumoconiosis se puede complicar con nódulos reumatoideos en pulmón, ésta asociación se llama Síndrome de Caplan. Posibles causas de muerte: cáncer, insuficiencia respiratoria, cor pulmonale.
Fibrosis pulmonar idiopática (Síndrome de Haman Rich)
Es más frecuente en varones adultos. Es un proceso destructivo de los tabiques alveolares de origen desconocido que lleva a la fibrosis intersticial difusa con el cuadro de pulmón en panal. El agente responsable no se ha podido identificar, pero produce episodios repetidos de lesión aguda que culminan en la fibrosis. Los linfocitos Th2 colaboran con la inflamación. Los neumocitos tipo I liberan TGF-beta, y este parece ser el motor del proceso, como estímulo fundamental de fibroblastos. La fibrosis es subpleural y sigue los septos interlobulillares. No hay cambios morfológicos más allá de la fibrosis. Por esto último, por se un proceso idiopático y porque puede verse en otros procesos, el diagnóstico se hace por descarte. Clínica: hipoxemia, cianosis grave, lleva a la muerte por insuficiencia respiratoria en 3 años. Tratamiento: trasplante de pulmón.
Neumonía organizativa criptogenética
Etiología desconocida, se producen formaciones fibrosas polipoideas dentro de alvéolos y bronquiolos (cuerpos de Masson), que en la radiografía se ven como áreas de condensación. No produce pulmón en panal.
Clínica: disnea, tos, se recuperan con corticoides en tratamiento durante 6 meses.
Fármacos, irradiación
Autoinmunes: lupus, artritis reumatoidea, esclerodermia
GRANULOMATOSAS
Neumonitis por hipersensibilidad o alveolitis alérgica
Se da por inhalación de polvos orgánicos, como antígenos de plumas, de excrementos de aves, de esporas bacterianas, de esporas de hongos.
· Patogenia: 6 hs. después de la esposición se produce una reacción de hipersensibilidad de tipo III, con depósito de complejos inmunes en las paredes de los pequeños vasos pulmonares del intersticio. También hay reacción de tipo IV, con formación de granulomas en intersticio.
· Morfología: hay inflamación intersticial, fibrosis, granulomas.
· Clínica: 6 hs. después de la exposición presentan fiebre, tos, disnea y leucocitosis.
· Ejemplos:
1. El pulmón del granjero: es por exposición al heno almacenado húmedo, donde proliferan actinomicetos.
2. El pulmón del cuidador de palomas: por inhalación de antígenos de plumas o excrementos de aves.
3. El pulmón del aire acondicionado: por bacterias termófilas presentes en agua caliente.
Sarcoidosis
EOSINOFÍLICAS
Eosinofilia pulmonar
El ejemplo es el Síndrome de Loeffler. Son áreas de condensación cambiantes de eosinófilos a nivel pulmonar, desencadenadas por la presencia de parásitos como Ascaris lumbricoides. Hay liberación aumentada de IL5.
POR TABACO
Neumonitis intersticial descamativa (NID)
Es más frecuente en varones entre 40 y 50 años, es rara.
· Morfología: hay aumento de macrófagos en los alvéolos, que contienen un pigmento pardo grisáceo (macrófago del fumador) con escasos neumocitos descamados, rara vez se produce una fibrosis intersticial difusa que lleva a la insuficiencia respiratoria. Buen pronóstico si abandonan el tabaco.
Enfermedad pulmonar intersticial asociada con bronquiolitis respiratoria
Macrófagos semejantes a la NID, a nivel de bronquiolos respiratorios, puede progresar hacia la fibrosis.
HEMORRÁGICAS
Síndrome de Goodpasture
Enfermedad autoinmune donde se producen anticuerpos contra la membrana basal glomerular y contra los tabiques alveolares (especificamente contra el colágeno de tipo 4, cadena alfa3). Hay predisposición genética en relación con los HLA. Es más frecuente en varones de 20 a 30 años.
· Clínica: produce hemoptisis y una glomerulonefritis rápidamente progresiva (esta última es la principal causa de muerte, lleva a insuficiencia renal).
· Tratamiento: inmunosupresores y plasmaféresis (recambio de plasma).
Vasculitis
OTRAS
Proteinosis alveolar pulmonar
Dentro de los alvéolos se acumula un material semejante al surfactante, es rico en proteínas y fosfolípidos. Se acompaña de macrófagos y algunos neumocitos necróticos.
· Patogenia: defecto en el aclaramiento de los macrófagos.
· Hay 3 variantes:
1. Adquirida (90%): se da en adultos, hay carencia del factor de crecimiento hematopoyético (GM-CSF) por autoanticuerpos contra el mismo. Generalmente no progresa hacia la fibrosis intersticial. Clínica: tos, disnea, esputo (material gelatinoso) que da riesgo de infecciones.
2. Congénita: donde hay déficit del factor o del receptor para el GM-CSF, o mutaciones en proteínas del surfactante. Lleva a la muerte 3 a 6 meses luego del nacimiento.
3. Secundaria: rara, se asocia a neoplasias, inmunodeficiencias.
PLEURA
PLEURITIS
· Serosa: Por infecciones virales, no dejan secuelas.
· Fibrinosa: por lupus, uremia, artritis reumatoidea, radioterapia. Se resuelve o se organiza.
· Supurada (empiema): es por la llegada de bacterias u hongos al espacio pleural. Las vías de llegada son desde un foco pulmonar, por vía hemática, o desde un absceso subdiafragmático. Generalmente se organizan, dejando secuelas graves (adherencias).
· Hemorrágica: se da por neoplasias pleurales, infecciones por rickettsias. Secuelas graves.
TUMORES
· Mesotelioma benigno o tumor fibroso solitario: es un fibroma, se lo considera un tumor de tejidos blandos, se conecta a la pleura por un pedículo. Células semejantes a fibroblastos, CD 34 +, queratina -.
· Mesotelioma maligno: se asocia al asbesto y produce derrame pleural. Se origina en pleura visceral o parietal. El VS40 (virus de los simios 40) podría tener relación (inactivaría al P53 y RB). Tienen 3 variantes:
1. Sarcomatoide o mesenquimal (20%): se parece a un fibrosarcoma, son células fusiformes.
2. Epitelial (60%): CD 34 -, queratina +. Forma papilas, es dificil distinguirlo de un adenocarcinoma bronquioloalveolar.
3. Mixto (20%): combinación de los 2 anteriores. Producen una afectación difusa. Invaden pulmón, ganglios regionales y metástasis al hígado. Clínica: dolor torácico, disnea, derrame pleural recurrente. Mueren al año del diagnóstico.
LARINGE
LARINGITIS
Se producen por:
· Crup: el bacilo diftérico produce una toxina que genera la formación de una seudomembrana en la mucosa laringea, ésta tiende a desprenderse, siendo grave en niños pequeños por el escaso calibre de la laringe, pudiendo llevar a la muerte por asfixia.
· Seudocrup: lo producen los virus, las secreciones serosas se depositan en las cuerdas vocales, se deshidratan y general una disnea inspiratoria con el estridor laringeo. La obstrucción generalmente es parcial y no llega a la asfixia.
TUMORES
· Polipos laringeos: nódulos redondos, sésiles o pediculados, miden hasta 1 cm., se ubican en las cuerdas vocales. Se asocian a tabaco, someter la voz a grandes esfuerzos (maestras, cantantes, se lo llama el nódulo de los cantantes). Están formados por un eje de tejido conectivo rodeado de epitelio escamoso. Clínica: ronquera progresiva. En los cantantes es bilateral, en el resto unilateral.
· Papiloma laringeo: se parece a una frambuesa, mide hasta 1 cm. Se ubica en las cuerdas vocales. Son varios ejes digitiformes de tejido conectivo rodeados de epitelio escamoso. Se producen por HPV 6 u 11. Si aparece en niños pequeños suele ser una papilomatosis, y el contagio se produjo por aspiración en el canal del parto desde lesiones de condoloma acuminado materno. Clínica: ronquera, si son múltiples puede obstruir la vía aérea.
· Carcinoma epidermoide (maligno, 95%): es más frecuente en varones mayores de 40 años. Predisponentes: tabaco, asbesto, infección por HPV, alcohol, irradiación. Surgen a partir de hiperplasias atípicas y displasia previa.
Morfología: pueden ser intrínsecos (crecen hacia el interior de la laringe) o extrínsecos (crecen hacia afuera, invadiendo estructuras vecinas). Según la ubicación pueden estar:
1. Por arriba de las cuerdas vocales, esto genera disfagia.
2. En las cuerdas vocales, son los más frecuentes y los que tienen bajo riesgo de metástasis porque están bien diferenciados y por la escasa presencia de linfáticos en las cuerdas. Generan disfonía.
3. Por debajo de las cuerdas vocales, generan disnea.
Evolución: metastatizan a ganglios regionales del cuello. La sobrevida a los 5 años es del 50%.
CAVIDADES NASALES Y SENOS PARANASALES. FARINGE
SINUSITIS
Es la inflamación de los senos paranasales. Surge de una rinitis complicada. La rinitis produce obstrucción de un meato, esto es una circunstancia de estasis, que predispone al ingreso de bacterias hacia el seno, cargándose de pus. Puede haber complicaciones, en el caso del seno maxilar puede alcanzar la órbita, el resto de los senos pueden producir osteomielitis y desde allí meningitis. El seno maxilar se puede comprometer a partir de una infección periapical desde la boca, en este caso aparece una flora infecciosa mixta proveniente de la misma. Existe una asociación poco frecuente con el Síndrome de Kartagener.
FARINGITIS
En el 90% son de origen viral (los mismos virus de las rinitis), en el 10% son de origen bacterianas por estafilococo y estreptococo beta hemolítico del grupo A (existe relación con fiebre reumatoidea y glomerulonefritis postestreptocócica).
TUMORES
· Angiofibroma nasofaringeo: se da en jóvenes, más frecuente en varones, formado por abundantes vasos, su característica es ser muy sangrante, pudiendo llevar a la muerte por la hemorragia.
· Papiloma invertido: crece al revés, hacia el hueso, infiltrando. Para curarlo hay que extirparlo completamente. Suele aislarse a los HPV 6 u 11. Histología: epitelio escamoso o cilíndrico. Otras variantes de papilomas son el septal (que es el más frecuente) y cilíndrico, ambas variantes son exofíticas.
· Carcinoma nasofaringeo (maligno): de células escamosas queratinizante, de células escamosas no queratinizante (EVB) e indiferenciado (EVB).
Se ubica más frecuentemente en el cavum (el techo de la faringe). Puede haber asociación con el EBV. Otros predisponentes son el tabaco y las nitrosaminas. En Africa son más frecuentes en niños, pero en China son más frecuentes en adultos. Diseminan a ganglios cervicales. La sobrevida a los 3 años es del 60%.
· Linfoma de células NK/T, de tipo nasal (granuloma letal de la línea media, maligno): tiene infecciones bacterianas añadidas con formación de granulomas y ulceraciones. Se creía que era una inflamación crónica de mal pronóstico por su penetración, hoy se sabe que es un linfoma.
· Neuroblastoma olfatorio (maligno): estesioneuroblastoma, se origina en las células neuroendócrinas dispersas (sinaptofisina, CD56, enolasa neuronal específica y cromogranina +) en la mucosa olfatoria. Son células redondas pequeñas semejantes a neuroblastos. Poseen gránulos secretores. Son ampliamente metastatizantes. Sobrevida a los 5 años del 60%.
· Plasmocitoma (o mieloma solitario): forma polipoide, generalmente no se transforma en mieloma múltiple.
miércoles, 7 de agosto de 2013
PATOLOGÍAS RESPIRATORIAS I: ANOMALÍAS CONGÉNITAS, EDEMA DE PULMON, ATELECTASIA, EPOC Y NEUMONÍAS
ANOMALÍAS CONGÉNITAS
Agenesia: es la falta de uno o los dos pulmones, siendo esto ultimo incompatible con la vida.
Hipoplasia pulmonar: poco desarrollo, en el caso de ser bilateral lleva a una insuficiencia respiratoria en la infancia. Puede transplantarse.
Secuestro pulmonar: un segmento del pulmón es irrigado solo por las bronquiales y no hay contacto con la vía aérea. Predispone a bronquiectasias. Es una circunstancia de estasis, fuera de la sangre puede haber infecciones. Ej.: hiperplasia de próstata (disminución de orina, no barre las bacterias y se estanca en vejiga).
Quistes congénitos: son fragmentos de intestino primitivo. Se dividen en broncogénicos (centrales, únicos miden hasta 5 cm., contienen mucina o aire, se recubren de una pared de epitelio respiratorio, pueden llevar a infecciones) y pulmonares (múltiples, periféricos pueden dar infecciones y al estar cercanos a la pleura, se pueden romper y dar neumotórax).
Hamartoma: tejido embrionario desorganizado.
EDEMA DE PULMÓN
La causa mas frecuente es la insuficiencia cardíaca izquierda. Otra causa es el SDRA (síndrome de distres respiratorio agudo).
· Patogenia: el ventrículo izquierdo es insuficiente, provocando congestión en las venas pulmonares, así aumenta la presión hidrostatica, y hay escape del liquido hacia el intersticio. Comienzan a actuar los linfáticos cuando se ven superados en su función el liquido se acumula y comienza a presionar a los alvéolos rompiendo su epitelio, y produce microhemorragias y edema intraalveolar.
· Morfología el liquido se acumula en las bases. Histología se ven macrofagos cargados de hemosiderina, que son las células de la insuficiencia cardíaca que pueden verse en el esputo.
· Clínica: tos con esputo color salmón y escamoso, disnea.
· Complicaciones: se pueden dar infecciones llevando al paciente a la muerte.




ENFERMEDADES PULMONARES OBSTRUCTIVAS CRÓNICAS (EPOC)
Son un conjunto de enfermedades que provocan disminución de la hematosis por obstrucción de las vías aéreas.



ENFISEMA
Agrandamiento permanente y anormal de los espacios aéreos situados mas allá de los bronquiolos terminales, acompañado de destrucción de sus paredes, sin fibrosis. Hay que distinguirlo de hiperinsuflacion, donde hay agrandamiento compensador, sin destrucción de las paredes.
Tipos:
Centrolobulillar: las partes proximales están afectadas, es mas frecuente en los lóbulos superiores y se asocia al tabaco y al polvo de carbón.
Panlobulillar: se afectan las porciones proximales y distales, es mas frecuente en lóbulos inferiores y se asocia al déficit genético de alfa1-Antitripsina.
Paraseptal: se afectan las partes distales, se asocia a áreas de cicatrización (TBC, sarcoidosis).
Irregular: no tienen un patrón definido, se asocia a procesos cicatrizales, es la variante morfológica mas frecuente pero generalmente no tiene importancia clínica.




Agenesia: es la falta de uno o los dos pulmones, siendo esto ultimo incompatible con la vida.
Hipoplasia pulmonar: poco desarrollo, en el caso de ser bilateral lleva a una insuficiencia respiratoria en la infancia. Puede transplantarse.
Secuestro pulmonar: un segmento del pulmón es irrigado solo por las bronquiales y no hay contacto con la vía aérea. Predispone a bronquiectasias. Es una circunstancia de estasis, fuera de la sangre puede haber infecciones. Ej.: hiperplasia de próstata (disminución de orina, no barre las bacterias y se estanca en vejiga).
Quistes congénitos: son fragmentos de intestino primitivo. Se dividen en broncogénicos (centrales, únicos miden hasta 5 cm., contienen mucina o aire, se recubren de una pared de epitelio respiratorio, pueden llevar a infecciones) y pulmonares (múltiples, periféricos pueden dar infecciones y al estar cercanos a la pleura, se pueden romper y dar neumotórax).
Hamartoma: tejido embrionario desorganizado.
EDEMA DE PULMÓN
La causa mas frecuente es la insuficiencia cardíaca izquierda. Otra causa es el SDRA (síndrome de distres respiratorio agudo).
· Patogenia: el ventrículo izquierdo es insuficiente, provocando congestión en las venas pulmonares, así aumenta la presión hidrostatica, y hay escape del liquido hacia el intersticio. Comienzan a actuar los linfáticos cuando se ven superados en su función el liquido se acumula y comienza a presionar a los alvéolos rompiendo su epitelio, y produce microhemorragias y edema intraalveolar.
· Morfología el liquido se acumula en las bases. Histología se ven macrofagos cargados de hemosiderina, que son las células de la insuficiencia cardíaca que pueden verse en el esputo.
· Clínica: tos con esputo color salmón y escamoso, disnea.
· Complicaciones: se pueden dar infecciones llevando al paciente a la muerte.
ATELECTASIA
Es la expansión incompleta de los pulmones con conservación de la irrigacion, es reversible.
Congénita: se ve en el SDR del recién nacido.
Adquirida: puede ser:
· Obstructiva: obstrucción completa de una vía aérea (por tumores, cuerpos extraños, bronquitis crónica). El mediastino se desplaza hacia el lugar de la atelectasia.
· Compresiva: es la mas frecuente, se produce por ocupación de la cavidad pleural con líquidos sangre, tumores, aire, etc.; también por elevación anormal del diafragma por peritonitis, abscesos y tumores subdiafragmaticos. El mediastino se desplaza hacia el lado contrario de la atelectasia.
· Por contracción se da por cambios cicatrizales que provocan retracción del parénquima pulmonar. No es reversible.
· Parcheadas: espacios con perdida de surfactante, se ven en el SDR.
Patología humana, Robbins, 8va edición
Patología humana, Robbins, 8va edición
Son un conjunto de enfermedades que provocan disminución de la hematosis por obstrucción de las vías aéreas.
Patología humana, Robbins, 8va edición
ENFISEMA
Agrandamiento permanente y anormal de los espacios aéreos situados mas allá de los bronquiolos terminales, acompañado de destrucción de sus paredes, sin fibrosis. Hay que distinguirlo de hiperinsuflacion, donde hay agrandamiento compensador, sin destrucción de las paredes.
Tipos:
Centrolobulillar: las partes proximales están afectadas, es mas frecuente en los lóbulos superiores y se asocia al tabaco y al polvo de carbón.
Panlobulillar: se afectan las porciones proximales y distales, es mas frecuente en lóbulos inferiores y se asocia al déficit genético de alfa1-Antitripsina.
Paraseptal: se afectan las partes distales, se asocia a áreas de cicatrización (TBC, sarcoidosis).
Irregular: no tienen un patrón definido, se asocia a procesos cicatrizales, es la variante morfológica mas frecuente pero generalmente no tiene importancia clínica.
Patología humana, Robbins, 8va edición
Es mas frecuente en varones entre 50 y 75 años (la panacinar se da mas en jóvenes).
Patología humana, Robbins, 8va edición
· Morfología: septos alveolares destruidos, con imágenes en palillo de tambor o en bastón El diagnostico de tipo morfológico se hace con secciones pulmonares gruesas, de 2 mm. de espesor (cortes de Gough) que se observan a simple vista o con lupa. También se observa inflamación en las pequeñas vías respiratorias, con neutrofilos, macrofagos, LT y LB.
Patología humana, Robbins, 8va edición
· Clínica: cuando aparecen los síntomas ya se ha perdido el 33% de la capacidad pulmonar. Los pacientes presentan disnea, tos seca, disminución de peso, tórax en tonel, espiración alargada, respiran con los labios apretados y la facies es rubicunda.
· Causas de muerte: core pulmonale, IRC, neumotórax (por ruptura de bullas: espacios enfisematosos mayores a 1 cm. de diámetro que al estar cercanos a la pleura pueden romperse).
Patología humana, Robbins, 8va edición
BRONQUITIS CRÓNICA
Paciente que presenta tos persistente con esputo, al menos 3 meses al año, en 2 años consecutivos. Es mas frecuente en varones después de los 40 años.
· Patogenia: se da por irritacion (principalmente el tabaco, tambien por silice, polvo de cereales) y por infecciones microbianas.
· Morfología hipertrofia de glándulas submucosas en traquea y bronquios, e hiperplasia de células caliciformes en bronquios y bronquiolos. El epitelio bronquial puede presentar zonas de metaplasia escamosa y displasia a causa del tabaco. En los bronquiolos hay tapones de moco. Hay infiltrado inflamatorio crónico en bronquios y bronquiolos.
· Clínica: tos con expectoración purulenta, facies cianotica.
· Causas de muerte: core pulmonale, IRC, cáncer de pulmón.



ASMA BRONQUIAL
Es el aumento de la irritabilidad del árbol traqueobronquial que produce un estrechamiento brusco de las vías aéreas bronquiales y puede revertirse espontáneamente o con tratamiento.




Tipos:
Extrínseca
· Alérgica: es la mas frecuente y se da por exposición a un alergeno como polen, acaro del polvo, caspa de animales. Produce reacción de hipersensibilidad de tipo I. Hay muchos genes implicados como HLA clase II, ADAM 33 (gen que codifica una metaloproteína que, en bronquios, aumenta la proliferación de musculo liso y fibroblastos).
Morfología: se forman tapones de moco que ocluyen bronquios y bronquiolos. Dentro de los tapones se observan: los espirales de Curshman, que son espirales de epitelio descamado, y los cristales de Charcot-Leyden, que son acumulos de proteínas de membrana del eosinofilo. Se ve también hipertrofia e hiperplasia del musculo liso bronquial por las broncoconstricciones e infiltrado de eosinofilos en la pared bronquial. Hay aumento de colágeno de tipo I y III en la membrana basal de la mucosa. Los eosinofilos liberan PBM (proteína básica mayor), con la cual rompen epitelio sano.
Clínica: disnea, sibilancias, alargamiento de la espiración (la dificultad esta en la salida de aire, porque queda atrapado dentro de los tapones mucosos).
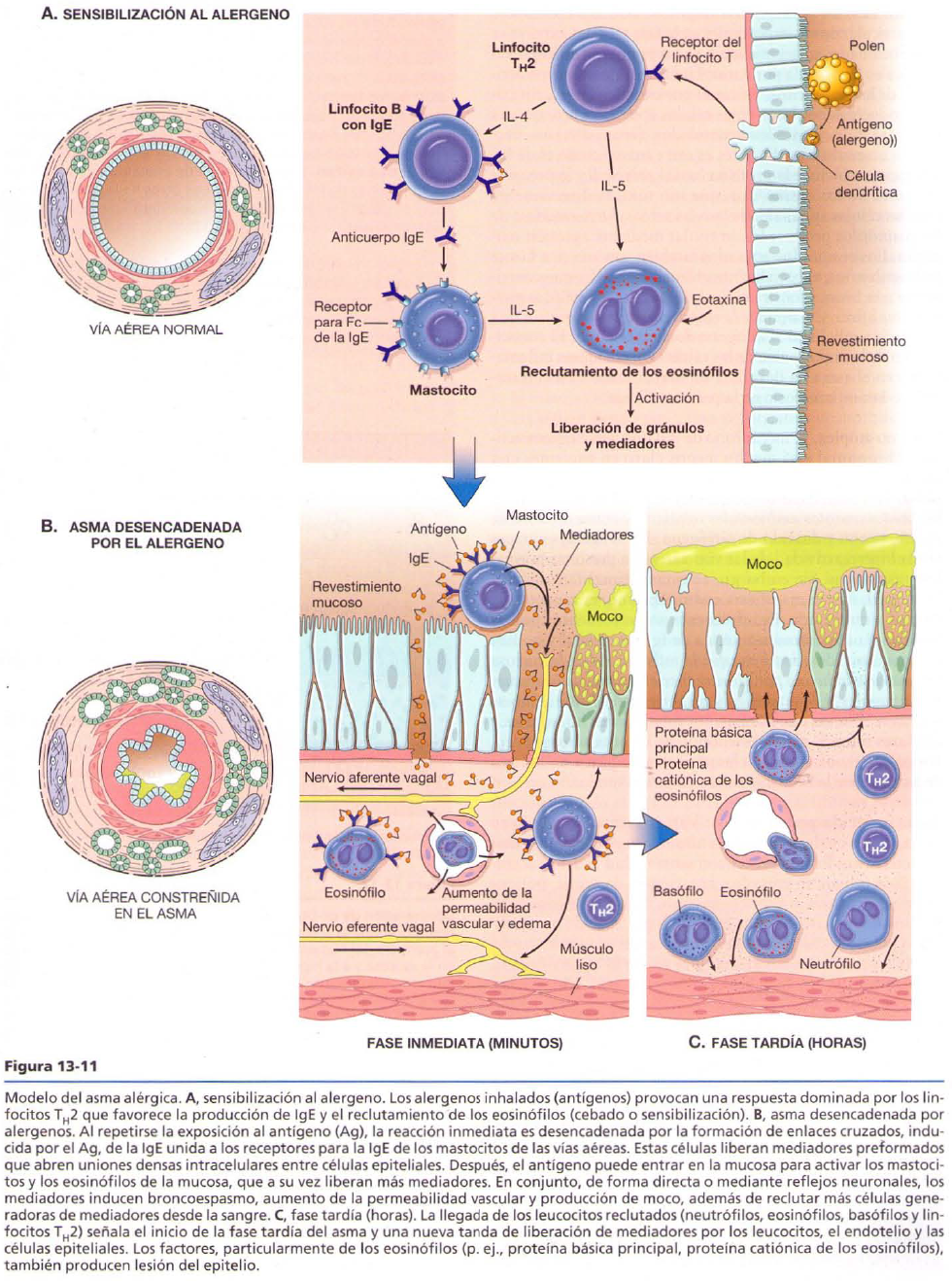
· Ocupacional: por inhalación de humos, gases químicos se produce primero una reacción de tipo I y luego de tipo III.
· Por aspergilosis: es por el Aspergillus fulmigatus. Se produce primero una reacción de tipo I y luego de tipo III.
Intrínseca
· Sensible a la aspirina: solo en pacientes susceptibles. La aspirina inhibe a la ciclooxigenasa, y en estos pacientes hay desvío hacia una mayor producción de leucotrienos.
· No atopica: se da por virus, frío estrés contaminantes ambientales como dióxido de azufre y dióxido de nitrógeno. Hay irritación vagal directa.

BRONQUIECTASIAS
Dilatación anormal y permanente de bronquios y bronquiolos, provocada por infecciones crónicas necrotizantes.
Causas:
· Congénitas fibrosis quistica, inmunodeficiencias congénitas, secuestro pulmonar, síndrome de Kartagener o de los cilios inmóviles: defecto de la movilidad ciliar (compromete cilias y el flagelo del espermatozoide), esta formado por sinusitis, bronquiectasias, infertilidad y situs inversus.
· Adquiridas: son mas frecuentes. Obstrucción (tumores, cuerpos extraños, bronquitis crónicas) e infecciones (secuelas de TBC, estafilococo, sarampión, HIV), y menos frecuentes son el lupus y la artritis reumatoidea.
Morfología: la afectación es bilateral a nivel de los lóbulos inferiores donde las vías aéreas están dilatadas hasta 4 veces el tamaño normal, se afectan las vías mas verticales. Esta dilatación puede ser cilíndrica fusiforme o sacular. La luz de los bronquios presenta pus, y la mucosa ulceras. La flora bacteriana es mixta e incluso a veces hongos como el Aspergillus fumigatus, la bacteria cae a la dilatación porque no hay mas cilias.
Clínica: tos con espectoracion maloliente, fiebre, a veces hemoptisis por debilidad de la pared que esta dilatada.
Complicaciones: core pulmonale, amiloidosis, abscesos.

· Patogenia: se da por irritacion (principalmente el tabaco, tambien por silice, polvo de cereales) y por infecciones microbianas.
· Morfología hipertrofia de glándulas submucosas en traquea y bronquios, e hiperplasia de células caliciformes en bronquios y bronquiolos. El epitelio bronquial puede presentar zonas de metaplasia escamosa y displasia a causa del tabaco. En los bronquiolos hay tapones de moco. Hay infiltrado inflamatorio crónico en bronquios y bronquiolos.
· Clínica: tos con expectoración purulenta, facies cianotica.
· Causas de muerte: core pulmonale, IRC, cáncer de pulmón.
Patología humana, Robbins, 8va edición
ASMA BRONQUIAL
Es el aumento de la irritabilidad del árbol traqueobronquial que produce un estrechamiento brusco de las vías aéreas bronquiales y puede revertirse espontáneamente o con tratamiento.
Tipos:
Extrínseca
· Alérgica: es la mas frecuente y se da por exposición a un alergeno como polen, acaro del polvo, caspa de animales. Produce reacción de hipersensibilidad de tipo I. Hay muchos genes implicados como HLA clase II, ADAM 33 (gen que codifica una metaloproteína que, en bronquios, aumenta la proliferación de musculo liso y fibroblastos).
Morfología: se forman tapones de moco que ocluyen bronquios y bronquiolos. Dentro de los tapones se observan: los espirales de Curshman, que son espirales de epitelio descamado, y los cristales de Charcot-Leyden, que son acumulos de proteínas de membrana del eosinofilo. Se ve también hipertrofia e hiperplasia del musculo liso bronquial por las broncoconstricciones e infiltrado de eosinofilos en la pared bronquial. Hay aumento de colágeno de tipo I y III en la membrana basal de la mucosa. Los eosinofilos liberan PBM (proteína básica mayor), con la cual rompen epitelio sano.
Clínica: disnea, sibilancias, alargamiento de la espiración (la dificultad esta en la salida de aire, porque queda atrapado dentro de los tapones mucosos).
Patología humana, Robbins, 8va edición
· Ocupacional: por inhalación de humos, gases químicos se produce primero una reacción de tipo I y luego de tipo III.
· Por aspergilosis: es por el Aspergillus fulmigatus. Se produce primero una reacción de tipo I y luego de tipo III.
Intrínseca
· Sensible a la aspirina: solo en pacientes susceptibles. La aspirina inhibe a la ciclooxigenasa, y en estos pacientes hay desvío hacia una mayor producción de leucotrienos.
· No atopica: se da por virus, frío estrés contaminantes ambientales como dióxido de azufre y dióxido de nitrógeno. Hay irritación vagal directa.
Patología humana, Robbins, 8va edición
BRONQUIECTASIAS
Dilatación anormal y permanente de bronquios y bronquiolos, provocada por infecciones crónicas necrotizantes.
Causas:
· Congénitas fibrosis quistica, inmunodeficiencias congénitas, secuestro pulmonar, síndrome de Kartagener o de los cilios inmóviles: defecto de la movilidad ciliar (compromete cilias y el flagelo del espermatozoide), esta formado por sinusitis, bronquiectasias, infertilidad y situs inversus.
· Adquiridas: son mas frecuentes. Obstrucción (tumores, cuerpos extraños, bronquitis crónicas) e infecciones (secuelas de TBC, estafilococo, sarampión, HIV), y menos frecuentes son el lupus y la artritis reumatoidea.
Morfología: la afectación es bilateral a nivel de los lóbulos inferiores donde las vías aéreas están dilatadas hasta 4 veces el tamaño normal, se afectan las vías mas verticales. Esta dilatación puede ser cilíndrica fusiforme o sacular. La luz de los bronquios presenta pus, y la mucosa ulceras. La flora bacteriana es mixta e incluso a veces hongos como el Aspergillus fumigatus, la bacteria cae a la dilatación porque no hay mas cilias.
Clínica: tos con espectoracion maloliente, fiebre, a veces hemoptisis por debilidad de la pared que esta dilatada.
Complicaciones: core pulmonale, amiloidosis, abscesos.
Patología humana, Robbins, 8va edición

NEUMONIAS (infecciones pulmonares)
Se producen cuando fracasan los mecanismos de defensa normales locales o la inmunidad del huésped. Por ejemplo: supresión del reflejo de la tos (coma, anestesia, fármacos , trastornos de las cilias (humo del tabaco, gases corrosivos, etc.), falla en el aclaramiento de los macrófagos (alcohol, tabaco), congestión del pulmón acumulo de secreciones por obstrucciones, inmunodeficiencias.


BACTERIANA (consolidativa)
Hacen focos de condensación ocupando los espacios alveolares. Según distribución se dividen en:
Bronconeumonía o neumonía broncolobulillar: se da en niños pequeños, ancianos e inmunocomprometidos.
· Los agentes etiológicos son variados: Neumococo, Estafilococo, E. coli, Haemófilus influenzae, Pseudomona aeruginosa, Klebsiella, Legionella pneumóphila, Estafilococo aureus, Moraxella catarrhalis.
· Morfología: la afectación es bilateral y bibasal. Son focos múltiples de consolidación, no mayores de 4 cm.
· Histología: los focos presentan exudado supurado que rellena principalmente las zonas de bronquiolos y alvéolos.
· Clínica: fiebre, rales, tos con espectoración.
· Complicaciones: abscesos, diseminación a la pleura (empiema), diseminación a pericardio (pericarditis supurada), bacteriemia (puede producir abscesos en otros órganos), organización del exudado.
Neumonía lobar: condensación de un lóbulo o parte del mismo. Se da en jóvenes y adolescentes, con más prevalencia en hombres.
· El agente etiológico que está presente en el 95% de los casos es el neumococo, sobretodo los serotipos 1, 2, 3 y 7, siendo el más virulento el 3 porque puede producir abscesos. En el 5% restante, son los agentes mencionados anteriormente.
· Patogenia: la puerta de entrada es el aparato respiratorio, y la patogenicidad del neumococo reside en su cápsula que inhibe la fagocitosis (no se adhieren las opsoninas). Los anticuerpos contra la cápsula confieren inmunidad.
· Morfología: atraviesa 4 etapas:
1. Congestión: dura 24 hs., hay escaso líquido seroso intraalveolar, escasos neutrófilos y elevado número de bacterias. Hay rales.
2. Hepatización roja: se produce la condensación de los espacios alveolares, el sector comprometido se parece a un tejido sólido como un hígado, de ahí el nombre de hepatización. Se llama roja porque junto con los neutrófilos extravasan eritrocitos que tiñen la condensación. El exudado es fibrinoso. No hay rales, hay silencio auscultatorio.
3. Hepatización gris: el exudado se vuelve fibrinopurulento. Los eritrocitos se desintegran, lo que cambia la coloración de la condensación. Hay silencio auscultatorio, matidez.
4. Resolución: sin tratamiento se suele presentar en 8 a 10 días. Se reabsorbe el exudado y se expulsa por la tos. Hay rales.
Con ATB se acorta la infección en 2 a 3 días. Hay vacuna para pacientes de riesgo.
· Clínica: escalofríos (que representan crisis de bacteriemias), fiebre muy alta, rales o silencio según la etapa. Si afecta la pleura, hay dolor en puntada de costado. En algunos casos se producen complicaciones.



ATÍPICA PRIMARIA (neumonitis)
Es intersticial. Se llama atípica porque produce una inflamación intersticial, y no hay focos de condensación. Se observa escaso exudado seroso intraalveolar e infiltrado de mononucleares (linfocitos) en intersticio.
· Los agentes son virales como el virus de la gripe (influenza A y B), rhinovirus, coxsakie. También puede darse por micoplasma (en este caso pueden producirse crioaglutininas y criohemolisinas) y chlamydias. Se llama primaria porque en cierto porcentaje de casos es de causa desconocida, aunque se sospecha viral (no se confirma por laboratorio).
· Clínica: es el típico cuadro gripal, con fiebre, dolores articulares, musculares, disnea, tos seca y escasos signos radiográficos. Si se complica con una infección bacteriana cambia el cuadro histológico y puede llevar a la muerte.
Otra forma grave es el SARS (síndrome respiratorio agudo grave): producido por un coronavirus, provocó grandes epidemias en los países asiáticos, con un 10% de mortalidad.
En el 2009 influenza A sufrió una mutación mayor a partir de genoma viral aviar, siendo de contagio humano (H5N1). Es el responsable de la pandemia actual de gripe. La mortalidad depende de la sobreinfección bacteriana o la posibilidad de cuadros hemorrágicos y diseminados (miocarditis).

ABSCESO PULMONAR
Acumulación localizada de pus, con necrosis licuefactiva del tejido pulmonar. Se produce por una siembra profunda de bacterias. Es más frecuente en adultos.
· Los agentes son el estafilococo, neumococo tipo 3 y bacterias anaerobias de la boca como peptococus, bacteroides, fusobacterium.
· Las vías de llegada son:
1. Aspiración de material infectivo (es la más frecuente), desde la boca, por aspiración del contenido gástrico, por cuerpos extraños.
2. A partir de procesos infecciosos pulmonares (bronquiectasias, neumonías).
3. Embolos sépticos.
4. Traumatismos penetrantes de pulmón.
5. Cáncer de pulmón (en el 10% de los abscesos hay un carcinoma oculto).
· Morfología: miden hasta 6 cm., si se abren hacia un bronquio se expulsa su contenido, quedando una cavidad residual. Esta cavidad puede contaminarse con bacterias u hongos, así adquiere un color verdinegro con olor fétido, cuadro llamado gangrena pulmonar.
· Clínica: expectoración maloliente purulenta, tos, fiebre.
· Complicaciones: se pueden liberar émbolos sépticos.
NEUMONÍAS EN EL INMUNODEPRIMIDO
En los pacientes con SIDA se produce infección por gérmenes oportunistas y es de alta mortalidad. Suele haber más de un germen a la vez. Los gérmenes más frecuentes son:
1. Bacterias: Pseudomona, Mycobacterium, Legionella y Listeria momocytogenes.
2. Virus: CMV y herpes.
3. Hongos: cándida, Cryptococcus neoformans, Aspergillus y Pneumocystis Jiroveci (antes llamado carinii).



NEUMONÍAS CRÓNICAS
La mayoría de las veces es una lesión localizada en un paciente inmunocompetente, con o sin afectación de los ganglios linfáticos regionales. Hay inflamación granulomatosa que se puede deber a bacterias u hongos.
Tuberculosis
Es una enfermedad granulomatosa crónica transmisible producida por Mycobacterium tuberculosis. Puede afectar a otros órganos. Los centros de los granulomas experimentan necrosis caseosa.
· Tuberculosis pulmonar primaria: complejo de Ghon, buena evolución o forma progresiva.
· Tuberculosis pulmonar secundaria: se reactiva pero en los vértices. Buena evolución o forma progresiva.
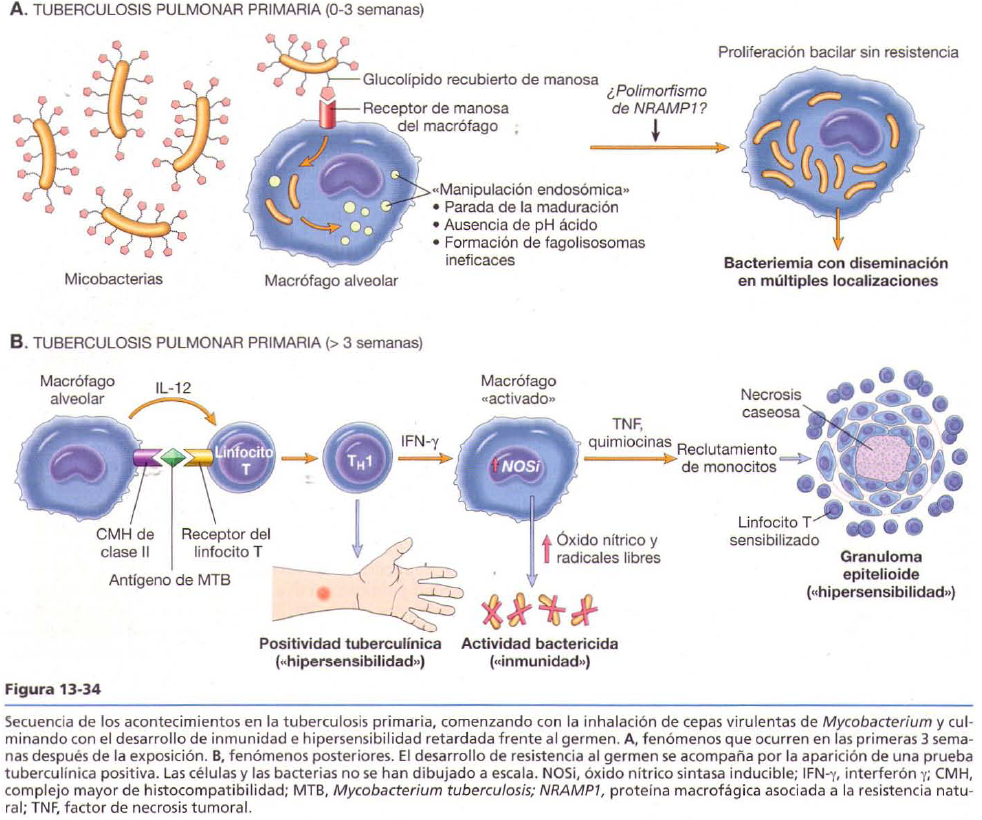





Enfermedad bacteriana no tuberculosa
Histoplasmosis, coccidioidomicosis y blastomicosis
Patología humana, Robbins, 8va edición
Clasificación según síndrome clínico:
· Neumonía aguda de la comunidad (Neumococo).
· Neumonía atípica de la comunidad (mycoplasma, chlamydias y virus).
· Neumonía nosocomial (Estafilococo aureus, Klebsiella, E. coli, Pseudomona).
· Neumonía por aspiración (flora anaerobia de la boca).
· Neumonía crónica (granulomatosas como TBC, hongos).
· Neumonías necrotizantes y abscesos (anaerobias de la boca, Estafilococo, Klebsiella, Neumococo tipo 3).
· Neumonía en inmunodeprimidos (virus, bacterias y hongos).
BACTERIANA (consolidativa)
Hacen focos de condensación ocupando los espacios alveolares. Según distribución se dividen en:
Bronconeumonía o neumonía broncolobulillar: se da en niños pequeños, ancianos e inmunocomprometidos.
· Los agentes etiológicos son variados: Neumococo, Estafilococo, E. coli, Haemófilus influenzae, Pseudomona aeruginosa, Klebsiella, Legionella pneumóphila, Estafilococo aureus, Moraxella catarrhalis.
· Morfología: la afectación es bilateral y bibasal. Son focos múltiples de consolidación, no mayores de 4 cm.
· Histología: los focos presentan exudado supurado que rellena principalmente las zonas de bronquiolos y alvéolos.
· Clínica: fiebre, rales, tos con espectoración.
· Complicaciones: abscesos, diseminación a la pleura (empiema), diseminación a pericardio (pericarditis supurada), bacteriemia (puede producir abscesos en otros órganos), organización del exudado.
Neumonía lobar: condensación de un lóbulo o parte del mismo. Se da en jóvenes y adolescentes, con más prevalencia en hombres.
· El agente etiológico que está presente en el 95% de los casos es el neumococo, sobretodo los serotipos 1, 2, 3 y 7, siendo el más virulento el 3 porque puede producir abscesos. En el 5% restante, son los agentes mencionados anteriormente.
· Patogenia: la puerta de entrada es el aparato respiratorio, y la patogenicidad del neumococo reside en su cápsula que inhibe la fagocitosis (no se adhieren las opsoninas). Los anticuerpos contra la cápsula confieren inmunidad.
· Morfología: atraviesa 4 etapas:
1. Congestión: dura 24 hs., hay escaso líquido seroso intraalveolar, escasos neutrófilos y elevado número de bacterias. Hay rales.
2. Hepatización roja: se produce la condensación de los espacios alveolares, el sector comprometido se parece a un tejido sólido como un hígado, de ahí el nombre de hepatización. Se llama roja porque junto con los neutrófilos extravasan eritrocitos que tiñen la condensación. El exudado es fibrinoso. No hay rales, hay silencio auscultatorio.
3. Hepatización gris: el exudado se vuelve fibrinopurulento. Los eritrocitos se desintegran, lo que cambia la coloración de la condensación. Hay silencio auscultatorio, matidez.
4. Resolución: sin tratamiento se suele presentar en 8 a 10 días. Se reabsorbe el exudado y se expulsa por la tos. Hay rales.
Con ATB se acorta la infección en 2 a 3 días. Hay vacuna para pacientes de riesgo.
· Clínica: escalofríos (que representan crisis de bacteriemias), fiebre muy alta, rales o silencio según la etapa. Si afecta la pleura, hay dolor en puntada de costado. En algunos casos se producen complicaciones.
Patología humana, Robbins, 8va edición
ATÍPICA PRIMARIA (neumonitis)
Es intersticial. Se llama atípica porque produce una inflamación intersticial, y no hay focos de condensación. Se observa escaso exudado seroso intraalveolar e infiltrado de mononucleares (linfocitos) en intersticio.
· Los agentes son virales como el virus de la gripe (influenza A y B), rhinovirus, coxsakie. También puede darse por micoplasma (en este caso pueden producirse crioaglutininas y criohemolisinas) y chlamydias. Se llama primaria porque en cierto porcentaje de casos es de causa desconocida, aunque se sospecha viral (no se confirma por laboratorio).
· Clínica: es el típico cuadro gripal, con fiebre, dolores articulares, musculares, disnea, tos seca y escasos signos radiográficos. Si se complica con una infección bacteriana cambia el cuadro histológico y puede llevar a la muerte.
Otra forma grave es el SARS (síndrome respiratorio agudo grave): producido por un coronavirus, provocó grandes epidemias en los países asiáticos, con un 10% de mortalidad.
En el 2009 influenza A sufrió una mutación mayor a partir de genoma viral aviar, siendo de contagio humano (H5N1). Es el responsable de la pandemia actual de gripe. La mortalidad depende de la sobreinfección bacteriana o la posibilidad de cuadros hemorrágicos y diseminados (miocarditis).
Patología humana, Robbins, 8va edición
ABSCESO PULMONAR
Acumulación localizada de pus, con necrosis licuefactiva del tejido pulmonar. Se produce por una siembra profunda de bacterias. Es más frecuente en adultos.
· Los agentes son el estafilococo, neumococo tipo 3 y bacterias anaerobias de la boca como peptococus, bacteroides, fusobacterium.
· Las vías de llegada son:
1. Aspiración de material infectivo (es la más frecuente), desde la boca, por aspiración del contenido gástrico, por cuerpos extraños.
2. A partir de procesos infecciosos pulmonares (bronquiectasias, neumonías).
3. Embolos sépticos.
4. Traumatismos penetrantes de pulmón.
5. Cáncer de pulmón (en el 10% de los abscesos hay un carcinoma oculto).
· Morfología: miden hasta 6 cm., si se abren hacia un bronquio se expulsa su contenido, quedando una cavidad residual. Esta cavidad puede contaminarse con bacterias u hongos, así adquiere un color verdinegro con olor fétido, cuadro llamado gangrena pulmonar.
· Clínica: expectoración maloliente purulenta, tos, fiebre.
· Complicaciones: se pueden liberar émbolos sépticos.
NEUMONÍAS EN EL INMUNODEPRIMIDO
En los pacientes con SIDA se produce infección por gérmenes oportunistas y es de alta mortalidad. Suele haber más de un germen a la vez. Los gérmenes más frecuentes son:
1. Bacterias: Pseudomona, Mycobacterium, Legionella y Listeria momocytogenes.
2. Virus: CMV y herpes.
3. Hongos: cándida, Cryptococcus neoformans, Aspergillus y Pneumocystis Jiroveci (antes llamado carinii).
Patología humana, Robbins, 8va edición
NEUMONÍAS CRÓNICAS
La mayoría de las veces es una lesión localizada en un paciente inmunocompetente, con o sin afectación de los ganglios linfáticos regionales. Hay inflamación granulomatosa que se puede deber a bacterias u hongos.
Tuberculosis
Es una enfermedad granulomatosa crónica transmisible producida por Mycobacterium tuberculosis. Puede afectar a otros órganos. Los centros de los granulomas experimentan necrosis caseosa.
· Tuberculosis pulmonar primaria: complejo de Ghon, buena evolución o forma progresiva.
· Tuberculosis pulmonar secundaria: se reactiva pero en los vértices. Buena evolución o forma progresiva.
Patología humana, Robbins, 8va edición
Enfermedad bacteriana no tuberculosa
Histoplasmosis, coccidioidomicosis y blastomicosis
Suscribirse a:
Comentarios (Atom)